

Umut Aydemir
I am a materials chemist having strong background in physics. I have an extensive experience in synthesis, chemical and physical characterization of novel functional materials focused on energy technologies.
36th International Conference on Thermoelectrics, Pasadena / California
July 30 - Aug 3, 2017
UPCOMING EVENTS
2017 MRS Spring Meeting & Exhibit
Phoenix, Arizona
April 17 - 21, 2017
MY LATEST RESEARCH

Complex multinary compounds (ternary, quaternary, and higher) offer countless opportunities for discovering new semiconductors for applications such as photovoltaics and thermoelectrics. However, controlling doping has been a major challenge in complex semiconductors as there are many possibilities for charged intrinsic defects (e.g., vacancies, interstitials, antisite defects) whose energy depends on competing impurity phases. Even in compounds with no apparent deviation from a stoichiometric nominal composition, such defects commonly lead to free carrier concentrations in excess of 1020 cm−3. Nevertheless, by slightly altering the nominal composition, these defect concentrations can be tuned with small variation of the chemical potentials (composition) of each element. While the variation of chemical composition is undetectable, it is shown that the changes can be inferred by mapping (in nominal composition space) the boundaries where different competing impurity phases form. In the inexpensive Zintl compound Ca9Zn4+xSb9, the carrier concentrations can be finely tuned within three different three-phase regions by altering the nominal composition (x = 0.2–0.8), enabling the doubling of thermoelectric performance (zT). Because of the low thermal conductivity, the zT can reach as high as 1.1 at 875 K, which is one of the highest among the earth abundant p-type thermoelectrics with no ion conducting.

The widespread use of thermoelectric conversion technology requires thermoelectric materials of high thermoelectric efficiency and high fracture strength. Single crystal SnSe shows an extremely high zT value in the moderate temperature range, but its mechanical properties have rarely been studied so far. Here we use density functional theory to determine the ideal strength and deformation mechanism of perfect SnSe single crystals for shear deformations. The lowest ideal strength of SnSe is found to be 0.59 GPa under the (100)/⟨001⟩ shear load, which agrees with experimental evidence that single crystals cleave along the (100) slip plane. The van der Waals-like Se−Sn bond, which couples the different Se−Sn layered substructures, is much softer than the covalent Se−Sn bond which constructs the Se−Sn layered substructure. This creates pathways of easy slip between Se−Sn layered substructures, which can release shear stress and lead to structural failure. Meanwhile, the layered substructures themselves can resist shearing within the (100)/⟨001⟩ slip system. These results provide a plausible atomic explanation for understanding the intrinsic mechanics of SnSe..

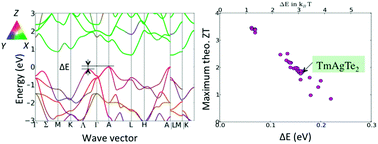
A new group of thermoelectric materials, trigonal and tetragonal XYZ2 (X, Y: rare earth or transition metals, Z: group VI elements), the prototype of which is TmAgTe2, is identified by means of high-throughput computational screening and experiment. Based on density functional theory calculations, this group of materials is predicted to attain high zT (i.e. ∼1.8 for p-type trigonal TmAgTe2 at 600 K). Among approximately 500 chemical variants of XYZ2 explored, many candidates with good stability and favorable electronic band structures (with high band degeneracy leading to high power factor) are presented. Trigonal TmAgTe2 has been synthesized and exhibits an extremely low measured thermal conductivity of 0.2–0.3 W m−1 K−1 for T > 600 K. The zT value achieved thus far for p-type trigonal TmAgTe2 is approximately 0.35, and is limited by a low hole concentration (∼1017 cm−3 at room temperature). Defect calculations indicate that TmAg antisite defects are very likely to form and act as hole killers. Further defect engineering to reduce such XY antisites is deemed important to optimize the zT value of the p-type TmAgTe2.
Thermoelectrics are gaining increasing interests due to their important potential applications. Many novel thermoelectric materials have been developed by manipulating the doping, electronic structure, phonon structure and scattering, as well as microstructure. Chemistry affects all of these enabling, for example, the tuning and engineering of the electron band structure or phonon scattering. This collection broadly covers the chemistry aspects of thermoelectric materials including their synthesis and processing, chemical and microstructure characterization, transport measurements and theory and modelling of their physical properties.
Intrinsically doped samples of YCuTe2 were prepared by solid state reaction of the elements. Based on the differential scanning calorimetry and the high temperature X-ray diffraction analyses, YCuTe2 exhibits a first order phase transition at ∼440 K from a low-temperature-phase crystallizing in the space group Pm1 to a high-temperature-phase in P. Above the phase transition temperature, partially ordered Cu atoms become completely disordered in the crystal structure. Small increases to the Cu content are observed to favour the formation of the high temperature phase. We find no indication of superionic Cu ions as for binary copper chalcogenides (e.g., Cu2Se or Cu2Te). All investigated samples exhibit very low thermal conductivities (as low as ∼0.5 W m−1 K−1 at 800 K) due to highly disordered Cu atoms. Electronic structure calculations are employed to better understand the high thermoelectric efficiency for YCuTe2. The maximum thermoelectric figure of merit, zT, is measured to be ∼0.75 at 780 K for Y0.96Cu1.08Te2, which is promising for mid-temperature thermoelectric applications.


Half-Heusler compounds based on XNiSn and XCoSb (X = Ti, Zr or Hf) have rapidly become important thermoelectric materials for converting waste heat into electricity. In this Review, we provide an overview on the electronic properties of half-Heusler compounds in an attempt to understand their basic structural chemistry and physical properties, and to guide their further development. Half-Heusler compounds can exhibit semiconducting transport behaviour even though they are described as ‘intermetallic’ compounds. Therefore, it is most useful to consider these systems as rigid-band semiconductors within the framework of Zintl (or valence-precise) compounds. These considerations aid our understanding of their properties, such as the bandgap and low hole mobility because of interstitial Ni defects in XNiSn. Understanding the structural and bonding characteristics, including the presence of defects, will help to develop different strategies to improve and design better half-Heusler thermoelectric materials.